Tutorial¶
This tutorial provides a workflow for creating and modifying SBML models. Basic Python knowledge is required. Modelers without programming experience could use command line scrips to convert SBML to spreadsheet and spreadsheet back to SBML, for this please check Command Line Scripts.
1. Convert SBML model to spreadsheet¶
As an example, download from BioModels database the SBML model BIOMD0000000010_url.xml. This is a kinetic SBML model based on a scientific publication from Kholodenko [MAPK].
Assuming the model is in the directory where you execute you Python code. This SBML model can be converted to a spreadsheet document as follows:
import sbmlxdf
model = sbmlxdf.Model('BIOMD0000000010_url.xml')
model.to_excel('BIOMD0000000010_ulr.xlsx')
You may now open the spreadsheet document. It will contain 5 sheets.
sbml
modelAttrs
unitDefs
compartments
species
reactions
Note
Number of sheets and their names in the spreadsheet document depend on the type of components stored in the model.
E.g. sheet sbml contains level and version of the SBML document, in this case Level 2 version 4.

reaction sheet (column names are highlighted)¶
The reaction sheet contains all information related to reactions this model:
id: identifier of the reaction
name: a more human readable name
metaid: required for miriamAnnotations and model history
miriamAnnotation: records (“;” - separated) with qualifier elements and resources (’http://identifiers.org/’ removed)
reversible: boolean value for reaction reversibility
reactants: reactants of the reaction (“;” - separated)
products
modifiers
kineticLaw
local parameters: parameters used in a kinetic law can be either local or global
reactionString: reaction converted into a tellurium compatible string representation
Note
SBML kinetics are change of amount per time unit and not change of concentration per time unit. In the kinetic laws you usually will pre-multiply the ‘traditional’ kinetic law with the compartment volume.
Note
Number of columns in a sheet depend on the type of attributes used
in the model. There are optional and mandatory attributes. Column order
does not matter, with the exception of the id column which must
be the first in most of the sheets (id it is used as an index).
2. Spreadsheet to SBML model¶
Continuing from above example, copy the spreadsheet and name it
BIO10_upd.xlsx. We are not changing anything in the model for now. We
just check how to convert a spreadsheet to SBML. We also verify that no
information gets lost during transfer and none of the attribute values change.
We convert the spreadsheet to SBML with following commands:
upd_model = sbmlxdf.Model('BIO10_upd.xlsx')
upd_model.export_sbml('BIO10_upd.xml')
BIO10_upd.xml should now be identical to the original SBML model
BIOMD0000000010_url.xml, at least it should contain the same information.
Note
Original and newly created SBML files are not exactly identical. sbmlxdf inserts a XML-comment with sbmlxdf creation time, libsbml version used and the model creation time. Sequence of XML elements and XML attributes might change.
To check if BIO10_upd.xml in fact contains all the information of the original
model, we can convert it back to spreadsheet and compare it with the
spreadsheet of the original model BIOMD0000000010_url.xlsx.
model = sbmlxdf.Model('BIO10_upd.xml')
model.export_sbml('BIO10_upd.xls')
Comparing BIOMD0000000010_url.xlsx with BIO10_upd.xlsx confirms,
that no information got lost in the transfer from spreadsheet to SBML and back
to spreadsheet.
3. Checking compliance with SBML specifications¶
Continuing from above. Though we successfully created a SBML model from a spreadsheet document, we have not checked if this model actually complies fully to SBML specifications. To improve model quality, it is recommended to first validate the model and only after successful validation create the SBML file.
libSBML, the API used in the background to access SBML data structures,
contains methods for checking compliance to SBML standards. These validation
routines are invoked with the method validate_sbml(). This method
creates an error report (tmp.txt) and a SBML file (tmp.xml), both in
directory ./results
upd_model = sbmlxdf.Model('BIO10_upd.xlsx')
upd_model.validate_sbml('tmp.xml')
{'Warnings': 10}
10 warnings are reported. Now open ./results/tmp.txt with a text editor.
first few lines of ./results/tmp.txt:
{'Warnings': 10} OK: SBML compliant
line 233: (99505 [Warning]) In situations where a mathematical expression
contains literal numbers or parameters whose units have not been declared,
is not possible to verify accurately the consistency of the units in the
expression.
The units of the <kineticLaw> <math> expression 'uVol * V1 * MKKK / ((1 +
pow(MAPK_PP / Ki, n)) * (K1 + MKKK))' cannot be fully checked. Unit
consistency reported as either no errors or further unit errors related to
this object may not be accurate.
line 294: (99505 [Warning]) In situations where a mathematical expression
contains literal numbers or parameters whose units have not been declared,
it is not possible to verify accurately the consistency of the units in the
expression.
The units of the <kineticLaw> <math> expression 'uVol * V2 * MKKK_P /
(KK2 + MKKK_P)' cannot be fully checked. Unit consistency reported as either
no errors or further unit errors related to this object may not be accurate.
line 345: (99505 [Warning]) In situations where a mathematical expression
contains literal numbers or parameters whose units have not been declared,
it is not possible to verify accurately the consistency of the units in the
expression.
These 10 warnings are all related to consistency of units of measurement. Line numbers in the report refer to related SBML document ‘./results/tmp.xml’.
While warnings are not critical and the SBML model could still be created,
here we try correcting the issues in the spreadsheet BIO10_upd.xlsx and
create a SBML model which passes the validation step, i.e. having correct
units of measurement.
4. Model Update¶
Continuing from above we’ll correct the warning messages in the spreadsheet
document BIO10_upd.xlsx. Also note that the SBML version is still at
Level 2 Version 4. Updates required for higher SBML versions would be more.
From the scientific paper [MAPK] we know that Michaelis constants are in the range of nanomolar (nM).
Let us try to introduce units into the model and also some other smaller modifications. This is actually an interactive step for the modeler. He will update the spreadsheet, import the spreadsheet with SBML and validate the model. In case of Warnings or Errors, further correction is required.
Finally following updates to the model remove any of the warning messages:
Additional unit definitions per_s, nM, nM_per_s in sheet unitDefs
(highlighted in green). metaid values were not specified as not required.

Updates to model reactions. We had to implement several configurations (updates in green).
Add unit definitions (which we defined in
unitsDefsheet) to all parameters inlocalParams. Unitless parameters, e.g.ngetdimensionlessassigned.kineticLawfor reactionJ0had a unitless1in the formula. We introduced a local parameterone, which has the unitdimensionlessassigned. Note: SBML L3V2 supports1 dimensionlessin the formula.We also modified the cryptic
metaids. This is not to correct any of the warnings. It rather demonstrates how we can change attributes to our liking.

In the sheet modelAttrs we could add the current time to the
modification history. We do this by adding ; localtime to the end of the
modifiedHistory value.

As there are no longer warnings, we can write out the model to SBML. Let’s give
it the name BIO10_L2V4_with_units.xml
upd_model = sbmlxdf.Model('BIO10_upd.xlsx')
upd_model.validate_sbml('tmp.xml')
{}
upd_model = sbmlxdf.Model('BIO10_L2V4_with_units.xml')
5. SBML model in latest SBML version¶
Continuing from above it is a small step to convert our model to the latest version of SBML, currently Level 3 version 2.
We just slightly update our spreadsheet BIO10_upd.xlsx, which is still in
Level 2 version 4.
First we have to change level and version in sheet sbml.

Validation check after this changes will show 30 warnings. Try it.
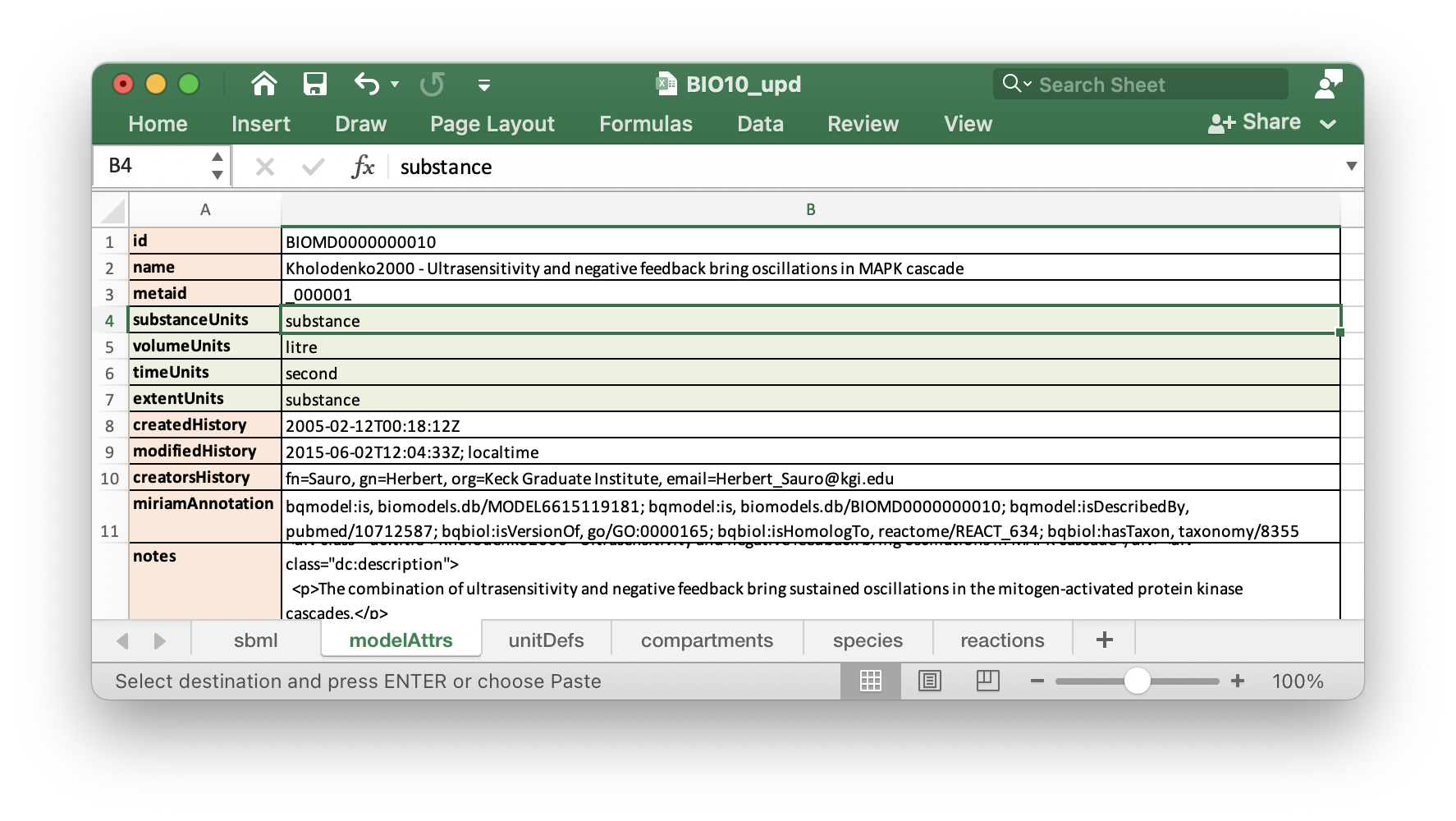
To get rid of these warnings, we have to add following attributes to sheet
modelAttrs:
substanceUnits
volumeUnits
timeUnits
extentUnits
In sheet reactions we should add stoic and const attributes
to reactants and products. constant is a mandatory attribute in
SBML L3V2.
With SBML L3V2 we also could change back the kinetics of reaction J0, i.e.
inserting 1 dimensionless into the kineticLaw.

Now we can generate from out spreadsheet a SBML L3V2 compliant model.
upd_model = sbmlxdf.Model('BIO10_upd.xlsx')
upd_model.validate_sbml('tmp.xml')
{}
upd_model = sbmlxdf.Model('BIO10_L3V2_with_units.xml')
6. Python access to SBML model data¶
Continuing from above we have generated from the SBML Biomodel
BIOMD0000000010_url.xml an updated SBML model in Level 3 Version 2 with
units of measurement added, BIO10_L3V2_with_units.xml.
Python programmers might require access to SBML data. Going through the pains
of interacting directly with libsbml can be avoided. sbmlxdf provides
access to SBML model data on the level of pandas dataframes.
You can extract SBML components and attributes, e.g. for use
in your optimization code. kineticLaws and other mathematical
constructs can be converted to Python functions, e.g. for ODE analysis.
To access SBML data, import a model and convert it to a set of dataframes:
model = sbmlxdf.Model('BIO10_L3V2_with_units.xml')
model_df = model.to_df()
print(model_df.keys())
dict_keys(['sbml', 'modelAttrs', 'unitDefs', 'compartments', 'species',
'reactions'])
Function sbmlxdf.extract_records() can be used to extract individual
records, e.g. there can be several reactants in a reaction.
Function sbmlxdf.extract_params() can be used to extract a dictionary of
key-value pairs from a record, e.g. to get all attributes of a reactant.
df_r = model_df['reactions']
print(len(df_r), 'reactions found, first reaction:' )
print(df_r.iloc[0])
print('\nreactants and products for some reactions:')
for id, reaction in df_r.head(3).iterrows():
print('reaction:', id)
for record in sbmlxdf.extract_records(reaction['reactants']):
print(' reactant: ', sbmlxdf.extract_params(record))
for record in sbmlxdf.extract_records(reaction['products']):
print(' product: ', sbmlxdf.extract_params(record))
10 reactions found, first reaction:
name MAPKKK activation
metaid J0
miriamAnnotation bqbiol:isHomologTo, reactome/REACT_525; bqbiol...
reversible False
reactants species=MKKK, stoic=1.0, const=True
products species=MKKK_P, stoic=1.0, const=True
modifiers species=MAPK_PP
kineticLaw uVol * V1 * MKKK / ((1 dimensionless + (MAPK_P...
localParams id=V1, value=2.5, units=nM_per_s; id=Ki, value...
Name: J0, dtype: object
reactants and products for some reactions:
reaction: J0
reactant: {'species': 'MKKK', 'stoic': '1.0', 'const': 'True'}
product: {'species': 'MKKK_P', 'stoic': '1.0', 'const': 'True'}
reaction: J1
reactant: {'species': 'MKKK_P', 'stoic': '1.0', 'const': 'True'}
product: {'species': 'MKKK', 'stoic': '1.0', 'const': 'True'}
reaction: J2
reactant: {'species': 'MKK', 'stoic': '1.0', 'const': 'True'}
product: {'species': 'MKK_P', 'stoic': '1.0', 'const': 'True'}
It is also possible to retrieve the stoichiometric matrix using:
print(model.get_s_matrix())
J0 J1 J2 J3 J4 J5 J6 J7 J8 J9
MKKK -1.0 1.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0
MKKK_P 1.0 -1.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0
MKK 0.0 0.0 -1.0 0.0 0.0 1.0 0.0 0.0 0.0 0.0
MKK_P 0.0 0.0 1.0 -1.0 1.0 -1.0 0.0 0.0 0.0 0.0
MKK_PP 0.0 0.0 0.0 1.0 -1.0 0.0 0.0 0.0 0.0 0.0
MAPK 0.0 0.0 0.0 0.0 0.0 0.0 -1.0 0.0 0.0 1.0
MAPK_P 0.0 0.0 0.0 0.0 0.0 0.0 1.0 -1.0 1.0 -1.0
MAPK_PP 0.0 0.0 0.0 0.0 0.0 0.0 0.0 1.0 -1.0 0.0
7. Run time course analysis¶
Once we have a SBML model, we might want to run a time course analysis to see the change of concentrations over time. Let us use the SBML model ‘BIO10_L3V2_with_units.xml’.
I’ll present two methods for time course analysis. Users without Python programming experience can use the graphical tool COPASI. Alternatively, you can use the Python package tellurium, but this requires some coding.
7.1 Time course analysis in COPASI¶
Assuming you already installed COPASI on your system. The example below is based on COPASI 4.33 (Build 246).
These are the steps for generating a time course plot:
open COPASI application
import the SBML model via menu
File -> Import SBML ..., select SBML fileBIO10_L3V2_with_units.xml. In my system a warning messages comes up, informing me that length unit and area unit are not defined. Don’t worry, these units are not used in our model.COPASI converts the SBML model into its internal format.

create a plot:
COPASI -> Output Specifications -> Plots[0]. Add aNewplot and add aNew Curve, select forX-AxisTime -> Model Time, select forY-AxisSpecies -> Transient Concentrationsand confirm.
preform time course analysis:
COPASI -> Tasks -> Time CourseSetDuration [s]to1800andIntervalsto1000. PressRun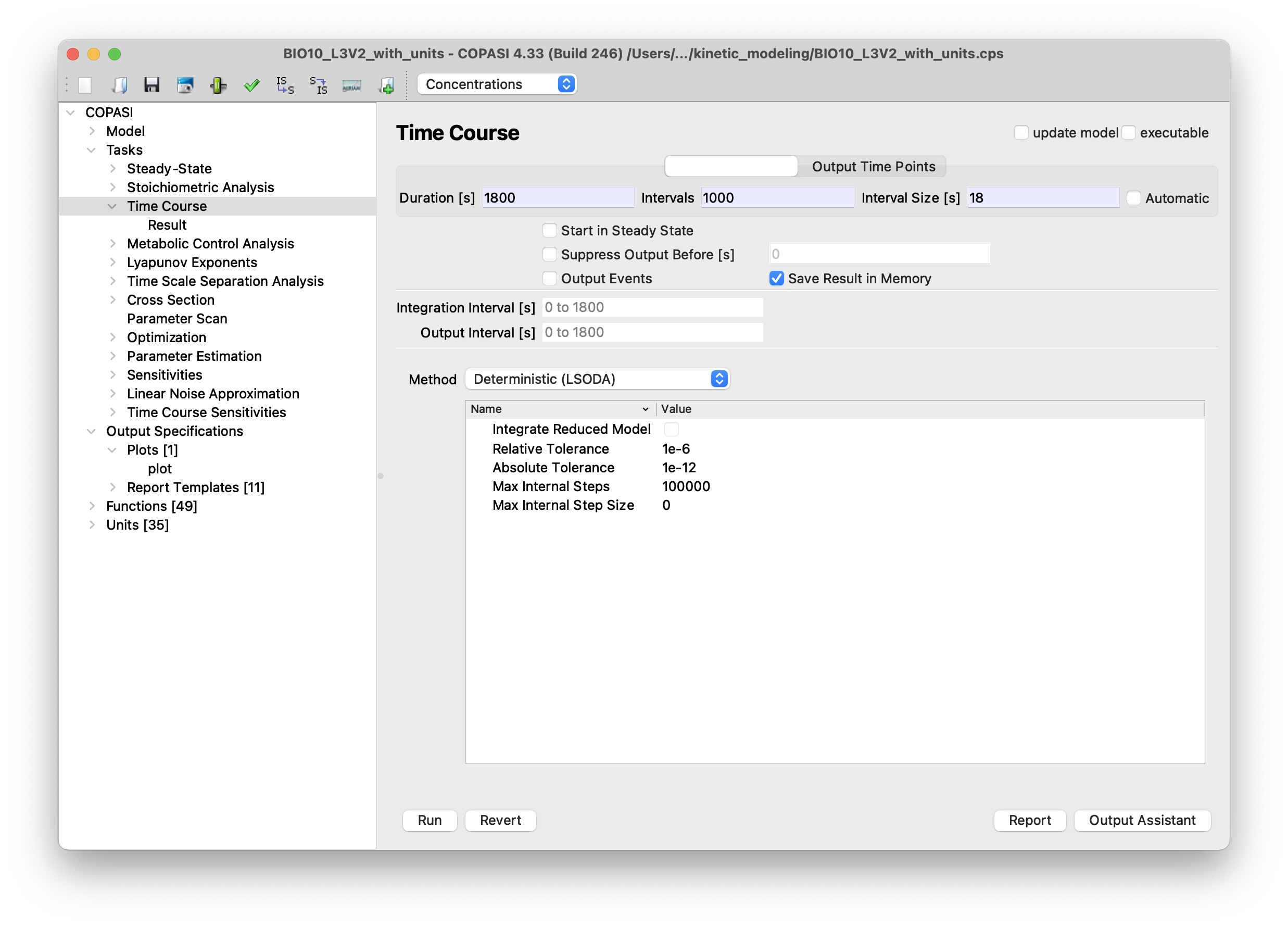
a plot showing the concentrations over time should appear. It not, the plot window might be hidden by other windows.

7.2 Time course in tellurium¶
Assuming you already pip installed the tellurium package.
import tellurium as te
r = te.loadSBMLModel('BIO10_L3V2_with_units.xml')
r.simulate(0, 1800, 1000)
r.plot()

8. Adding application specific annotations¶
As an implementer of optimization methods you might require application specific attributes on SBML components. This can be done using the annotation mechanism provided by SBML.
Continuing from the model above, we might be interested in the molecular
weight of the species involved in the reactions and possibly the length of the
proteins. We might required these attributes to configure additional
constraints during optimization. We could add this information as SBML
parameters to the parameters sheet (not discussed yet).
However, we would have difficulties linking species
objects with corresponding parameters object.
Using the annotation mechanism we can directly add our required attributes
on the level of species. Let us extend our spreadsheet BIO10_upd.xlsx.
To benefit from spreadsheet features, we
add a sheet helpers and copy relevant information from the
species sheet. The miriamAnnotation values provide references
to UniProt database. We manually lookup
UniProt and search for the unitprot ids used
in our model. In the Sequence section we find protein length and
molecular weight, which we add to our table.
Just as an example, we also add weights of inorganic phosphates.
Using Excel concat() function we create xmlAnnotations
using fixed namespace, token and prefix and
variable values from our table. Note: Here I define an arbitrary namespace.

Subsequently we copy the xmlAnnotation column and
Paste Special - Values it into our species sheet.
I also updated the metaid values.

We import the spreadsheet, validate it and create our new SBML
BIO10_L3V2_with_XML_annot.xml.
upd_model = sbmlxdf.Model('BIO10_upd.xlsx')
upd_model.validate_sbml('tmp.xml')
{}
upd_model.export_sbml('BIO10_L3V2_with_XML_annot.xml')
As a Python programmer you now have access to the additional attributes, which is demonstrated in below code block:
model = sbmlxdf.Model('BIO10_L3V2_with_XML_annot.xml')
model_df = model.to_df()
df_s = model_df['species']
for species, row in df_s.iterrows():
attrs = sbmlxdf.misc.extract_xml_attrs(row['xmlAnnotation'], token='molecule')
print(species, attrs)
MKKK {'weight_Da': '71960', 'prot_len': '638'}
MKKK_P {'weight_Da': '72039', 'prot_len': '638'}
MKK {'weight_Da': '43900', 'prot_len': '395'}
MKK_P {'weight_Da': '43821', 'prot_len': '395'}
MKK_PP {'weight_Da': '43900', 'prot_len': '395'}
MAPK {'weight_Da': '41257', 'prot_len': '361'}
MAPK_P {'weight_Da': '41336', 'prot_len': '361'}
MAPK_PP {'weight_Da': '41415', 'prot_len': '361'}